
Ab initio accelerated molecular dynamics study of the hydride ligands in the ruthenium complex: Ru(H2)2H2(P(C5H9)3)2†

Abstract
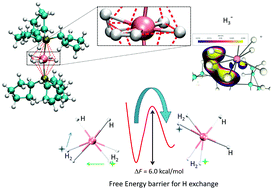
The dihydrogen complex Ru(H2)2H2(P(C5H9)3)2 has been investigated, via ab initio accelerated molecular dynamics, to elucidate the H ligands dynamics and possible reaction paths for H2/H exchange. We have characterized the free energy landscape associated with the H atoms positional exchange around the Ru centre. From the free energy landscape, we have been able to estimate a barrier of 6 kcal mol−1 for the H2/H exchange process. We have also observed a trihydrogen intermediate as a passing state along some of the possible reaction pathways.
 Please wait while we load your content...
Please wait while we load your content...

