
Formation mechanism and spectroscopy of C6H radicals in extreme environments: a theoretical study†

Abstract
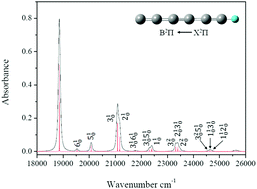
This study examined the reaction mechanisms of singlet (rhombic) and triplet (linear) C4 with acetylene by using accurate ab initio CCSD(T)/CBS//B3LYP/6-311G(d,p) calculations followed by a kinetic analysis of various reaction pathways and computations of relative product yields in combustion and planetary atmospheres. These calculations were combined with the Rice–Ramsperger–Kassel–Marcus (RRKM) calculations of reaction rate constants for predicting product-branching ratios, which depend on the collision energy under single-collision conditions. The results demonstrate that the initial reaction begins with the formation of an intermediate 3i2 with an entrance barrier of 3.0 kcal mol−1 and an intermediate 1i1 without entrance barriers. The product-branching ratios obtained by solving kinetic equations with individual rate constants calculated using the RRKM and variational transition-state theories for determining the collision energies between 5 kcal mol−1 and 25 kcal mol−1 demonstrate that l-C6H + H is the dominant reaction product, whereas HC3C3 + H, l-C6 + H2, c-C6H + H, and c-C6 + H2 are minor products. The electronic absorption spectra of solid neon matrices in the range of 17 140–22 200 cm−1 were obtained by Maier et al., and the optimized ground and excited state structures of C6H were used to simulate the absorption spectra by one-photon excitation equations. The displaced harmonic oscillator approximation and the Franck–Condon approximation were used to simulate the absorption spectrum of the B2Π ← X2Π transition of C6H. This indicates that the vibronic structures were dominated by one of the six active completely symmetric modes, with v3 being the most crucial.
 Please wait while we load your content...
Please wait while we load your content...

