
Statistically representative databases for density functional theory via data science†
 a
and
Roberto
Peverati
*a
a
and
Roberto
Peverati
*a
Abstract
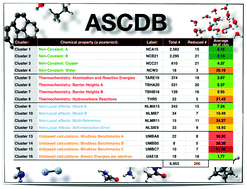
The amount of data and number of databases for the assessment and parameterization of density functional theory methods has grown substantially in the past two decades. In this work, we introduce a novel cluster analysis technique for density functional theory calculations of the electronic structure of atoms and molecules with the goal of creating new statistically significant databases with broad chemical scope, and a manageable number of data-points. By analyzing without a priori chemical assumptions a population of almost 350k data-points, we create a new database called ASCDB containing only 200 data-points. This new database holds the same chemical information as the larger population of data from which it is obtained, but with a computational cost that is reduced by several orders of magnitude. The labelling of the significant chemical properties is performed a posteriori on the resulting 16 subsets, classifying them into four areas of chemical importance: non-covalent interactions, thermochemistry, non-local effects, and unbiased calculations. The analysis of the results and their transferability shows that ASCDB is capable of providing the same information as that of the larger collection of data—such as GMTKN55, MGCDB84, and Minnesota 2015B—for several density functional theory methods and basis sets. In light of these results, we suggest the use of this new small database as a first inexpensive tool for the evaluation and parameterization of electronic structure theory methods.
 Please wait while we load your content...
Please wait while we load your content...

