
Stacking interactions of borazine: important stacking at large horizontal displacements and dihydrogen bonding governed by electrostatic potentials of borazine†
 ab
Snežana D.
Zarić
*ab
ab
Snežana D.
Zarić
*ab
Abstract
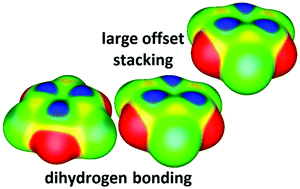
Potential energy surfaces of borazine–benzene and borazine–borazine stacking interactions were studied by performing DFT, CCSD(T)/CBS and SAPT calculations. The strongest borazine–benzene stacking was found in a parallel-displaced geometry, with a CCSD(T)/CBS interaction energy of −3.46 kcal mol−1. The strongest borazine–borazine stacking has a sandwich geometry, with a CCSD(T)/CBS interaction energy of −3.57 kcal mol−1. The study showed that borazine forms significant stacking interactions at large horizontal displacements (over 4.5 Å), with energies of −2.20 kcal mol−1 for the borazine–benzene and −1.96 kcal mol−1 for the borazine–borazine system. The strength of interactions and their geometrical preferences can be rationalized by observing the electrostatic potentials of borazine and benzene, which is in agreement with SAPT analysis showing that electrostatics is the most important energy component for borazine stacking. All the interactions found in crystal structures of borazine and related compounds were identified either as potential curve minima or the geometries obtained from their optimizations. We also report a new dihydrogen bonding dimer with a CCSD(T)/CBS interaction energy of −2.37 kcal mol−1, which is encountered in the borazine crystal structures and enables the formation of additional simultaneous interactions that contribute to the overall stability of the crystals.
 Please wait while we load your content...
Please wait while we load your content...

