
A charge optimized many-body potential for iron/iron-fluoride systems†
 b
and
J.
Mejía-López
*a
b
and
J.
Mejía-López
*a
Abstract
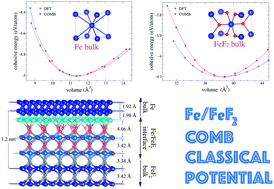
A classical interatomic potential for iron/iron-fluoride systems is developed in the framework of the charge optimized many-body (COMB) potential. This interatomic potential takes into consideration the effects of charge transfer and many-body interactions depending on the chemical environment. The potential is fitted to a training set composed of both experimental and ab initio results of the cohesive energies of several Fe and FeF2 crystal phases, the two fluorine molecules F2 and the F2−1 dissociation energy curve, the Fe and FeF2 lattice parameters of the ground state crystalline phase, and the elastic constants of the body centered cubic Fe structure. The potential is tested in an NVT ensemble for different initial structural configurations as the crystal ground state phases, F2 molecules, iron clusters, and iron nanospheres. In particular, we model the FeF2/Fe bilayer and multilayer interfaces, as well as a system of square FeF2 nanowires immersed in an iron solid. It has been shown that there exists a reordering of the atomic positions for F and Fe atoms at the interface zone; this rearrangement leads to an increase in the charge transfer among the atoms that make the interface and put forward a possible mechanism of the exchange bias origin based on asymmetric electric charge transfer in the different spin channels.
 Please wait while we load your content...
Please wait while we load your content...

