
Calculations of NO reduction with CO over a Cu1/PMA single-atom catalyst: a study of surface oxygen species, active sites, and the reaction mechanism†
 *ab
*ab
Abstract
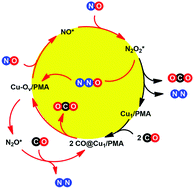
Density functional theory (DFT) calculations have been employed to probe the reaction mechanism of NO reduction with CO over a Cu1/PMA (PMA is the phosphomolybdate, Cs3PMo12O40) single-atom catalyst (SAC). Several important aspects of the catalytic system were addressed, including the generation of oxygen vacancies (Ov), formation of N2O2 intermediates, scission of the N–O bond of N2O2 intermediates to form N2O or N2, and decomposition of N2O to form N2. Unlike most previous theoretical studies, which tend to explore the reaction mechanism of polyoxometalate (POM) systems based on the isolated anionic unit, here, we build a model of the catalytic system with neutral species by introduction of counter cations to model the solid structure of the Cu1/PMA SAC. The major findings of our present study are: (1) CO adsorption on Cu sites leads to the formation of cationic Cu carbonyl species; (2) the Oc atom at the surface of the PMA support can easily react with the adsorbed CO to generate a Cu–Ov pair; (3) the Cu–Ov pair embedded on PMA is found to be the active site, not only for the formation of N2O2* by the reaction of two NO molecules via an Eley–Rideal pathway but also for the decomposition of N2O to form N2; (4) the adsorption of a NO molecule on the Cu–Ov pair with a bridging model results in charge transfer from the Cu atom to the π* antibonding orbital of the NO molecule; (5) IR spectroscopy of the key intermediates has been identified based on our DFT calculations; and (6) the Cu atom serves as an electron acceptor in Ov formation steps and an electron donor in N2O2 decomposition steps, and thus represents an electron reservoir. These results suggest that the POM-supported SAC with the cheaper Cu element is an efficient catalyst for the reaction between CO and NO.
 Please wait while we load your content...
Please wait while we load your content...

