
The influence of molecular geometry on the efficiency of thermally activated delayed fluorescence†
 ‡b
Zhongjie
Ren,
bd
Przemyslaw
Data,
ae
Andrei S.
Batsanov,
b
Thomas J.
Penfold,
*c
Martin R.
Bryce
*b
and
Fernando B.
Dias
*a
‡b
Zhongjie
Ren,
bd
Przemyslaw
Data,
ae
Andrei S.
Batsanov,
b
Thomas J.
Penfold,
*c
Martin R.
Bryce
*b
and
Fernando B.
Dias
*a
Abstract
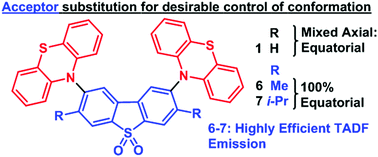
In this work we successfully developed a strategy for positively influencing the conformation of thermally activated delayed fluorescence (TADF) molecules containing phenothiazine as the electron donor (D) unit, and dibenzothiophene-S,S-dioxide as the acceptor (A), linked in D–A and D–A–D structures. In this strategy the effect of restricted molecular geometry is explored to maximize TADF emission. The presence of bulky substituents in different positions on the donor unit forces the molecules to adopt an axial conformer where the singlet charge transfer state is shifted to higher energy, resulting in the oscillator strength and luminescence efficiency decreasing. With bulky substituents on the acceptor unit, the molecules adopt an equatorial geometry, where the donor and acceptor units are locked in relative near-orthogonal geometry. In this case the individual signatures of the donor and acceptor units are evident in the absorption spectra, demonstrating that the substituent in the acceptor uncouples the electronic linkage between the donor and acceptor more effectively than with donor substitution. In contrast with the axial conformers that show very weak TADF, even with a small singlet triplet gap, molecules with equatorial geometry show stronger oscillator strength and luminescence efficiency and are excellent TADF emitters. Acceptor-substituted molecules 6 and 7 in particular show extremely high TADF efficiency in solution and solid film, even with a singlet–triplet energy gap around 0.2 eV. This extensive study provides important criteria for the design of novel TADF and room temperature phosphorescence (RTP) emitters with optimized geometry.
- This article is part of the themed collection: Functional Organic Materials for Optoelectronic Applications
 Please wait while we load your content...
Please wait while we load your content...

