
Predicting the preferred morphology of hexagonal boron nitride domain structure on nickel from ReaxFF-based molecular dynamics simulations†

Abstract
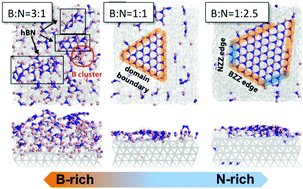
An understanding of the nucleation and growth of hexagonal boron nitride (hBN) on nickel substrates is essential to its development as a functional material. In particular, fundamental insights into the formation of the hexagonal lattices with alternating boron (B) and nitrogen (N) atoms could be exploited to control hBN lattice morphologies for targeted applications. In this study, the preferred shapes and edge configurations of atomically smooth hBN on Ni(111) were investigated using molecular dynamics (MD) simulations, along with reactive force field (ReaxFF) developed to represent the Ni/B/N system and the lattice-building B–N bond formation. The obtained hBN lattices, from different B : N feed ratios, are able to confirm that hBN domain geometries can indeed be tuned by varying thermodynamic parameters (i.e., chemical potentials of N and B) – a finding that has only been predicted using quantum mechanical theories. Here, we also showed that the nitrogen fed to the system plays a more crucial role in dictating the size of hBN lattices. With an increase of the relative N content, the simulated hBN domain shapes also transition from equilateral triangles to hexagons, again, consistent with the anticipation based on Density Functional Theory (DFT) calculations. Hence, a plausible approach to acquire a desired hBN nanostructure depends on careful control over the synthesis conditions, which now can benefit from reliable molecular simulations.
 Please wait while we load your content...
Please wait while we load your content...

