
Benchmarking DFT approaches for the calculation of polarizability inputs for refractive index predictions in organic polymers†
 *a
and
Johannes
Hachmann
*abc
*a
and
Johannes
Hachmann
*abc
Abstract
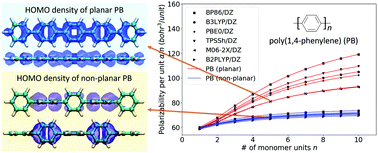
In a previous study, we introduced a new computational protocol to accurately predict the index of refraction (RI) of organic polymers using a combination of first-principles and data modeling. This protocol is based on the Lorentz–Lorenz equation and involves the calculation of static polarizabilities and number densities of oligomer sequences, which are extrapolated to the polymer limit. We chose to compute the polarizabilities within the density functional theory (DFT) framework using the PBE0/def2-TZVP-D3 model chemistry. While this ad hoc choice proved remarkably successful, it is also relatively expensive from a computational perspective. It represents the bottleneck step in the overall RI modeling protocol, thus limiting its utility for virtual high-throughput screening studies, in which efficiency is essential. For polymers that exhibit late-onset extensivity, the employed linear extrapolation scheme can require demanding calculations on long-oligomer sequences, thus becoming another bottleneck. In the work presented here, we benchmark DFT model chemistries to identify approaches that optimize the balance between accuracy and efficiency for this application domain. We compare results for conjugated and non-conjugated polymers, augment our original extrapolation approach with a non-linear option, analyze how the polarizability errors propagate into the RI predictions, and offer guidance for method selection.
 Please wait while we load your content...
Please wait while we load your content...

