
Ligand dynamics and protonation preferences of Rh and Ir complexes bearing an almost, but not quite, pendent base†
 a
W. E.
Christman,
a
J. Z.
Williams,
a
N.
Arulsamy,
a
A.
Goroncy
a
and
E. B.
Hulley
*a
a
W. E.
Christman,
a
J. Z.
Williams,
a
N.
Arulsamy,
a
A.
Goroncy
a
and
E. B.
Hulley
*a
Abstract
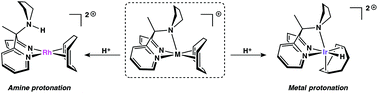
Pendent nucleophiles are essential partners in the cleavage and formation of bonds with hydrogen (e.g. protonation/deprotonation), but binding of the pendent group to the metal and the potential trapping of complexes in inactive states are a significant problem. The dipyridylmethane-based ligand framework bis(2-pyridyl)-N-pyrrolidinomethane (R,pyrCPy2), bearing a hemilabile pyrrolidine moiety, has been synthesized and complexes of the type [(R,pyrCPy2)M(COD)]X (COD = 1,5-cyclooctadiene) were prepared. The solution-phase ligand dynamics and relative protonation preferences were investigated via1H NMR spectroscopy; although favorable, pendent amine binding does not kinetically inhibit pendent base protonation. Protonation at the metal (with concomitant pyrrolidine binding) has been found to be favorable for Ir, whereas N-protonation is favorable for Rh. DFT calculations predict that the RhIII hydrides have much higher relative acidities than their Ir congeners (ΔpKa ≃ 7–8 in CH2Cl2), and are also more acidic than the strong acid [H(OEt2)2][B(C6F5)4].
 Please wait while we load your content...
Please wait while we load your content...

