
DFT modelling of a diphosphane − N-heterocyclic carbene–Rh(i) pincer complex rearrangement: a computational evaluation of the electronic effects in C–P bond activation†

Abstract
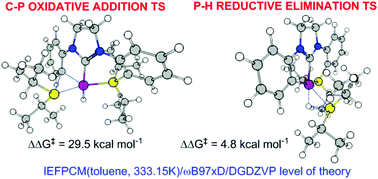
DFT calculations confirmed that the rearrangement of a PCP-Rh-H pincer to a CCP-Rh-phosphane pincer occured by C–P oxidative addition (ΔG‡ = 29.5 kcal mol−1, rate-determining step), followed by P–H reductive elimination (ΔG‡ = 4.8 kcal mol−1). The oxidative addition proceeded via a 3-centered transition state and is accelerated by electron-withdrawing substituents p- to the reacting C–P bond, resulting in a reaction constant (ρ) of 2.12 for ΔG‡ and 2.76 for ΔH‡ in a Hammett-type linear free energy relationship. AIM wavefunction analyses indicated a decrease in the negative charge on the carbon bonded to Rh with a concomitant increase in the positive charge on the latter. The electronic density at the Rh–P bond critical point and the atomic charge on Rh correlate well with the Hammett constants (σ) of the p-substituents. The replacement of the Rh-bound hydride with other anions (CH3, Ph, t-Bu, OH, F, Cl, and CN) results in a decrease in the OA barrier only for CH3, which is in accordance with the experimental results. The reductive elimination occurs via a 3-centered (Rh, H, P) transition state, which adopts a conformation wherein the steric clash between the i-Pr groups is minimized, followed by recomplexation of Rh and the newly formed (i-Pr)2PH by a conformational twist around the Rh–P axis.
 Please wait while we load your content...
Please wait while we load your content...

