
Electronic spectrum and characterization of diabatic potential energy surfaces for thiophenol†
 ab
Donald G.
Truhlar
*b
and
ab
Donald G.
Truhlar
*b
and
Abstract
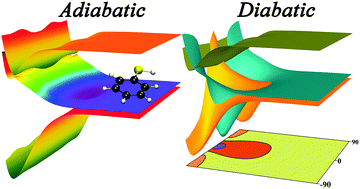
The electronic spectrum of thiophenol was simulated by a normal-mode sampling approach combined with TDDFT in the Tamm–Dancoff approximation (TDA). The vertical excitation energies were compared with electronic structure calculations by completely renormalized equation-of-motion coupled cluster theory with single and double excitations and noniterative inclusion of connected triples (CR-EOM-CCSD(T)) and by multi-reference perturbation theory. The spectrum was computed both with and without solvation effects, and these spectra are compared to each other and to experiment. Using multireference-perturbation-theory adiabatic wave functions and model-space diabatization by the fourfold way, diabatic potential energy surfaces of the lowest three singlet states (1ππ, 1ππ*, and 1nπσ*) were constructed along the S–H stretching coordinate, the C–C–S–H torsion coordinate, and the v16a and v16b normal coordinates. The first two of these two are primary coordinates for the photodissociation, and the diabatic crossing seams of the three states were calculated and plotted as functions of the two coordinates. The other two coordinates are out-of-plane ring distortion modes studied to assess the extent of their role in coupling the states near the first conical intersection, and the v16a mode was shown to be an important coupling mode there. The current study is the first step toward a detailed mechanistic analysis of the photoinduced S–H fission process of thiophenol, a test system to understand 1nπσ*-mediated reactions, at the same time already providing a better understanding of the thiophenol electronic excitations by clarifying the assignment of the experimental results.
 Please wait while we load your content...
Please wait while we load your content...

