
The importance of grand-canonical quantum mechanical methods to describe the effect of electrode potential on the stability of intermediates involved in both electrochemical CO2 reduction and hydrogen evolution†

Abstract
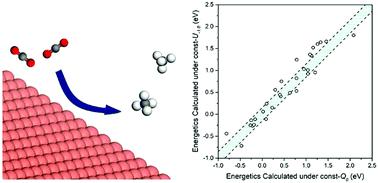
The rational design of electrocatalysts to convert CO2 to fuel requires predicting the effect of the electrode potential (U) on the binding and structures of the intermediates involved in CO2 electrochemical reduction (CO2ER). In this study, we used grand-canonical quantum mechanics (GC-QM) to keep the potential constant during the reactions (rather than keeping the charge constant as in standard QM) to investigate the effect of U on adsorption free energies (ΔGs) of 14 CO2ER intermediates on Cu(111) as well as the intermediates involved in the competitive hydrogen evolution reaction (HER). In contrast to most previous theoretical studies where ΔGs were calculated under constant charge (= 0, neutral), we calculated ΔGs under constant potential (U = 0.0, −0.5, −1.0, and −1.5 VSHE). By comparing the ΔGs calculated under constant U (= 0.0 VSHE) to those calculated under constant charge, we found differences up to 0.22 eV which would change the rates at 298 K by a factor of about 5300. In particular we found that the adsorption of species with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional group (i.e., *COOH, *CO, and *CHO) strengthened by up to 0.16 eV as U became more negative by 1 V, whereas the adsorption of –O– species (i.e., *OH, *OCH3, *COH, and *CHOH) weakened by up to 0.20 eV. For the (111) index surfaces of Cu, Au, Ag, Ir, Ni, Pd, Pt and Rh, we investigated the effect of U on the reaction free energy (ΔG) at pH = 0 for the crucial elementary steps for CO2ER (*CO + (H+/e−) → *CHO, ΔG = (ΔG*CHO − ΔG*CO) + eU) and HER (* + (H+/e−) → *H, ΔG = ΔG*H + eU. Our results indicated that the influence of U on (ΔG*CHO − ΔG*CO) was metal dependent. In contrast, the energy for converting a proton in solution to H* on the surface, ΔG*H, was barely affected by U (for the studied metals). Overall we found substantial differences (MAD > 0.18 eV) between the ΔGs calculated under U = −1.0 VSHE (relevant to experiments) and those calculated under constant charge (= 0, neutral) common to most theoretical investigations. Therefore, we strongly recommend application GC-QM to obtain accurate energetics for CO2ER.
O functional group (i.e., *COOH, *CO, and *CHO) strengthened by up to 0.16 eV as U became more negative by 1 V, whereas the adsorption of –O– species (i.e., *OH, *OCH3, *COH, and *CHOH) weakened by up to 0.20 eV. For the (111) index surfaces of Cu, Au, Ag, Ir, Ni, Pd, Pt and Rh, we investigated the effect of U on the reaction free energy (ΔG) at pH = 0 for the crucial elementary steps for CO2ER (*CO + (H+/e−) → *CHO, ΔG = (ΔG*CHO − ΔG*CO) + eU) and HER (* + (H+/e−) → *H, ΔG = ΔG*H + eU. Our results indicated that the influence of U on (ΔG*CHO − ΔG*CO) was metal dependent. In contrast, the energy for converting a proton in solution to H* on the surface, ΔG*H, was barely affected by U (for the studied metals). Overall we found substantial differences (MAD > 0.18 eV) between the ΔGs calculated under U = −1.0 VSHE (relevant to experiments) and those calculated under constant charge (= 0, neutral) common to most theoretical investigations. Therefore, we strongly recommend application GC-QM to obtain accurate energetics for CO2ER.
 Please wait while we load your content...
Please wait while we load your content...

