
Theoretical investigation of the infrared spectrum of small polyynes†
 *a
*a
Abstract
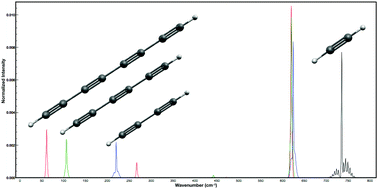
The full cubic and semidiagonal quartic force fields of acetylene (C2H2), diacetylene (C4H2), triacetylene (C6H2), and tetraacetylene (C8H2) are determined using CCSD(T) (coupled cluster theory with single and double excitations and augmented by a perturbative treatment of triple excitations) in combination with the atomic natural orbital (ANO) basis sets. Application of second-order vibrational perturbation theory (VPT2) results in vibrational frequencies that agree well with the known fundamental and combination band experimental frequencies of acetylene, diacetylene, and triacetylene (average discrepancies are less than 10 cm−1). Furthermore, the predicted ground state rotational constants (B0) and vibration–rotation interaction constants (αi) are shown to be consistent with known experimental values. New vibrational frequencies and rotational parameters from the presented theoretical predictions are given for triacetylene and tetraacetylene, which can be used to aid laboratory and astronomical spectroscopic searches for characteristic transitions of these molecules.
- This article is part of the themed collection: Theory, experiment, and simulations in laboratory astrochemistry
 Please wait while we load your content...
Please wait while we load your content...

