
Exploring adsorption of neutral aromatic pollutants onto graphene nanomaterials via molecular dynamics simulations and theoretical linear solvation energy relationships†

Abstract
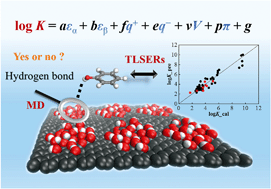
Predicting adsorption of organic pollutants onto graphene nanomaterials is not only useful for exploring their potential adsorbent applications, but also helpful for better understanding their fate and risks in aquatic environments. Herein molecular dynamics (MD) simulations and theoretical linear solvation energy relationships (TLSERs) were employed to construct prediction models for adsorption of neutral organic pollutants onto graphene and graphene oxides. The MD simulations for adsorption of 43 aromatic compounds onto graphene and diverse models of graphene oxides with various functional groups (hydroxyl, epoxy and carbonyl) demonstrate that graphene has a stronger affinity for the aromatic compounds than graphene oxides. The hydroxyl and carbonyl groups of graphene oxides were found to form hydrogen bonds with the aromatic adsorbates, while epoxy groups did not. TLSER models were developed for predicting the adsorption equilibrium coefficients (K) onto graphene and graphene oxide nanosheets. In the graphene prediction model, H-donating ability (εα) and dispersion/hydrophobic interactions (V) have significant effects on log K values, while in the graphene oxide model, εα is the most influential factor on log K values. The models provide in silico approaches for predicting adsorption affinities onto graphenic nanomaterials.
 Please wait while we load your content...
Please wait while we load your content...

