
Hybrid DFT investigation of the energetics of Mg ion diffusion in α-MoO3†

Abstract
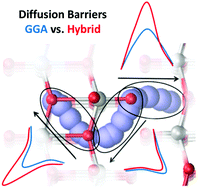
Rechargeable batteries that utilize divalent Mg ions as the charge carrier species can in principle achieve substantially greater volumetric energy densities than conventional Li-ion batteries. One significant impediment to the development of commercially viable Mg-ion batteries is the slow rate of Mg ion diffusion through otherwise promising cathode materials. Accurate prediction of the activation energies associated with this diffusion process using density functional theory (DFT) is especially challenging due to self-interaction errors intrinsic to DFT that lead to over-delocalization of the d-electrons. One effective but highly computationally demanding approach to reducing self-interaction errors is the use of hybrid functionals, which incorporate a fraction of exact Hartree–Fock exchange. In this work, we assess the effects of exact exchange on computed activation energies for ion diffusion in one potential cathode material, α-MoO3. In contrast to previous studies that primarily utilize non-hybrid functionals, we perform nudged elastic band calculations in which the nuclear coordinates are fully converged using both hybrid functionals and k-point sampling. It is found that while non-hybrid functionals indicate the existence of thermodynamically accessible channels for bulk Mg ion diffusion in all three dimensions, hybrid functionals predict that some of these channels are largely inaccessible under typical charge/discharge conditions. Furthermore, it is demonstrated that certain commonly used approximations for incorporating the effects of Hartree–Fock exchange are inadequate for this system, including DFT+U calculations and the use of single-point hybrid calculations using atomic positions obtained using non-hybrid functionals.
 Please wait while we load your content...
Please wait while we load your content...

