
Pseudorotaxanes in the gas phase: structure and energetics of protonated dibenzylamine–crown ether complexes†

Abstract
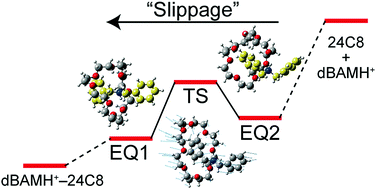
We observe UV spectra of protonated dibenzylamine (dBAMH+) and its complexes with 15-crown-5 (dBAMH+–15C5), 18-crown-6 (dBAMH+–18C6), and 24-crown-8 (dBAMH+–24C8) under cold (∼10 K) gas-phase conditions by UV photodissociation (UVPD) and UV–UV hole-burning (HB) spectroscopy. The UVPD spectrum of the dBAMH+–15C5 complex shows an extensive low-frequency progression, which originates from a unique conformation of the dBAMH+ part with benzene rings facing closely to each other, while UVPD and calculation results suggest open conformations of the dBAMH+ part for dBAMH+–18C6 and dBAMH+–24C8. UV–UV HB spectra of the dBAMH+–24C8 complex indicate that there exist at least two conformers; multiple conformations can contribute to high stability of dBAMH+–24C8 pseudorotaxane due to “conformational” entropic effects. The UVPD experiment indicates that the dissociation probability of dBAMH+–24C8 into dBAMH+ and 24C8 is substantially smaller than that of dBAMH+–15C5 and dBAMH+–18C6, which can be related to the barrier height in the dissociation process. The energetics of the dBAMH+–24C8 complex is investigated experimentally with NMR spectroscopy and theoretically with the global reaction route mapping (GRRM) method. An energy barrier of ∼60 kJ mol−1 is present in the pseudorotaxane formation in solution, whereas there is no barrier in the gas phase. In the course of the photodissociation, excited dBAMH+–24C8 complexes can be trapped at many local minima corresponding to multiple conformations. This can result in effective dissipation of internal energy into degrees of freedom not correlated to the dissociation and decrease the dissociation probability for the dBAMH+–24C8 complex in the gas phase. The energy barrier for the pseudorotaxane formation in solution originates not simply from the slippage process but rather from solvent effects on the dBAMH+–24C8 complex.
 Please wait while we load your content...
Please wait while we load your content...

