
Evaluating excited state atomic polarizabilities of chromophores†

Abstract
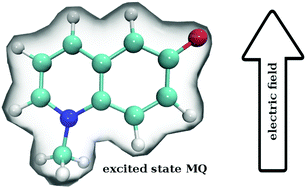
Ground and excited state dipoles and polarizabilities of the chromophores N-methyl-6-oxyquinolinium betaine (MQ) and coumarin 153 (C153) in solution have been evaluated using time-dependent density functional theory (TD-DFT). A method for determining the atomic polarizabilities has been developed; the molecular dipole has been decomposed into atomic charge transfer and polarizability terms, and variation in the presence of an electric field has been used to evaluate atomic polarizabilities. On excitation, MQ undergoes very site-specific changes in polarizability while C153 shows significantly less variation. We also conclude that MQ cannot be adequately described by standard atomic polarizabilities based on atomic number and hybridization state. Changes in the molecular polarizability of MQ (on excitation) are not representative of the local site-specific changes in atomic polarizability, thus the overall molecular polarizability ratio 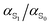 does not provide a good approximation for local atom-specific polarizability changes on excitation. Accurate excited state force fields are needed for computer simulation of solvation dynamics. The chromophores considered in this study are often used as molecular probes. The methods and data reported here can be used for the construction of polarizable ground and excited state force fields. Atomic and molecular polarizabilities (ground and excited states) have been evaluated over a range of functionals and basis sets. Different mechanisms for including solvation effects have been examined; using a polarizable continuum model, explicit solvation and via sampling of clusters extracted from a MD simulation. A range of different solvents have also been considered.
does not provide a good approximation for local atom-specific polarizability changes on excitation. Accurate excited state force fields are needed for computer simulation of solvation dynamics. The chromophores considered in this study are often used as molecular probes. The methods and data reported here can be used for the construction of polarizable ground and excited state force fields. Atomic and molecular polarizabilities (ground and excited states) have been evaluated over a range of functionals and basis sets. Different mechanisms for including solvation effects have been examined; using a polarizable continuum model, explicit solvation and via sampling of clusters extracted from a MD simulation. A range of different solvents have also been considered.
- This article is part of the themed collection: 2018 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

