
Theoretical characterization of sulfur-to-selenium substitution in an emissive RNA alphabet: impact on H-bonding potential and photophysical properties†

Abstract
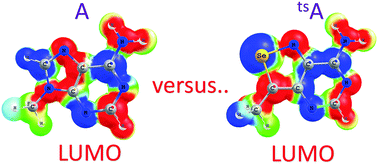
We employ density functional theory (DFT) and time-dependent DFT (TDDFT) calculations to investigate the structural, energetic and optical properties of a new computationally designed RNA alphabet, where the nucleobases, tsA, tsG, tsC, and tsU (ts-bases), have been derived by replacing sulfur with selenium in the previously reported tz-bases, based on the isothiazolo[4,3-d]pyrimidine heterocycle core. We find out that the modeled non-natural bases have minimal impact on the geometry and energetics of the classical Watson–Crick base pairs, thus potentially mimicking the natural bases in a RNA duplex in terms of H-bonding. In contrast, our calculations indicate that H-bonded base pairs involving the Hoogsteen edge of purines are destabilized as compared to their natural counterparts. We also focus on the photophysical properties of the non-natural bases and correlate their absorption/emission peaks to the strong impact of the modification on the energy of the lowest unoccupied molecular orbital. It is indeed stabilized by roughly 1.1–1.6 eV as compared to the natural analogues, resulting in a reduction of the gap between the highest occupied and the lowest unoccupied molecular orbital from 5.3–5.5 eV in the natural bases to 3.9–4.2 eV in the modified ones, with a consequent bathochromic shift in the absorption and emission spectra. Overall, our analysis clearly indicates that the newly modelled ts-bases are expected to exhibit better fluorescent properties as compared to the previously reported tz-bases, while retaining similar H-bonding properties. In addition, we show that a new RNA alphabet based on size-extended benzo-homologated ts-bases can also form stable Watson–Crick base pairs with the natural complementary nucleobases.
 Please wait while we load your content...
Please wait while we load your content...

