
Towards reliable references for electron paramagnetic resonance parameters based on quantum chemistry: the case of verdazyl radicals†

Abstract
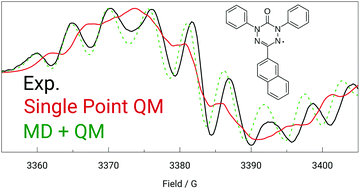
We present an efficient and accurate computational procedure to calculate properties measurable by EPR spectroscopy. We simulate a molecular dynamics (MD) trajectory by employing the quantum mechanically derived force field (QMDFF) [S. Grimme, J. Chem. Theory Comput., 2014, 10, 4497] and sample the trajectory at different time steps. For each snapshot EPR properties are calculated with a hybrid density functional theory (DFT) method. EPR spectra are simulated based on the averaged results. We applied the strategy to a number of previously published and novel verdazyl radicals, for which we recorded EPR spectra. The resulting simulated spectra are compatible with experiment already before employing an additional fitting step, in contrast to those from single point electronic-structure calculations. After the refinement, the experimental data are excellently reproduced, and the fitted EPR parameters do not deviate much from the calculated ones. This provides confidence in ascribing a direct physical meaning to the refined data in terms of experimental EPR parameters rather than merely considering them as mathematical fit parameters. We also find that couplings to hydrogen nuclei have a significant influence on the spectra of verdazyl radicals.
 Please wait while we load your content...
Please wait while we load your content...

