
Comparative density functional theory–density functional tight binding study of fullerene derivatives: effects due to fullerene size, addends, and crystallinity on band structure, charge transport and optical properties†

Abstract
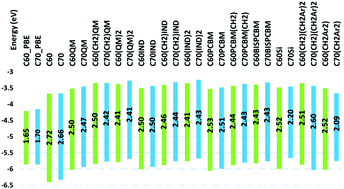
We present a systematic comparative density functional theory–density functional tight binding study of multiple derivatives of C60 and C70 with different addends, in molecular as well as solid state. In particular, effects due to fullerene size, type and number of addends, and of crystallinity on band structure, charge transport, and optical properties are investigated. These are important, in particular, for rational selection of fullerene derivatives as acceptor and electron transport layers in organic as well as planar inverted perovskite solar cells. We find that by the choice of type and number of addends, one can modulate the LUMO within 0.4 eV. Changes in the HOMO can reach 0.7 eV. Substituting C70 for C60 results in destabilization of the HOMO by about 0.1 eV for indene and quinodimethane addends and by a less significant amount for PCBM addends. The effect of C70–C60 substitution on the LUMO is of similar magnitude. A more significant change in HOMO–LUMO energy is seen for the aryl addends. On the other hand, all C70 based molecules have strong visible absorption. For most addends, the crystal packing leads to a stabilization of both the LUMO and HOMO by about ∼0.2 and ∼0.1 eV, respectively, vs. single molecules. When using bis-addends, it is also possible to enhance the visible absorption. Electron and hole transport rates are computed to vary vastly depending on the addends chosen; specifically, we compute that indene and quimodimethane addends can enhance charge transport rates while the aryl addend is predicted to result in substantially smaller mobilities of electrons and holes, vs. PC60BM. Furthermore, the –CH2 and bisaddend addition can significantly enhance the charge transfer rates for the PCBM addend.
 Please wait while we load your content...
Please wait while we load your content...

