
Predictive models of gas sorption in a metal–organic framework with open-metal sites and small pore sizes†

Abstract
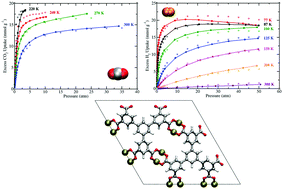
Simulations of CO2 and H2 sorption were performed in UTSA-20, a metal–organic framework (MOF) having zyg topology and composed of Cu2+ ions coordinated to 3,3′,3′′,5,5′,5′′-benzene-1,3,5-triyl-hexabenzoate (BHB) linkers. Previous experimental studies have shown that this MOF displays remarkable CO2 sorption properties and exhibits one of the highest gravimetric H2 uptakes at 77 K/1.0 atm (2.9 wt%) [Z. Guo, et al. Angew. Chem., Int. Ed., 2011, 50, 3178–3181]. For both sorbates, the simulations were executed with the inclusion of explicit many-body polarization interactions, which was necessary to reproduce sorption onto the open-metal sites. Non-polarizable potentials were also utilized for simulations of CO2 sorption as a control. The simulated excess sorption isotherms for both CO2 and H2 are in very good agreement with the corresponding experimental data over a wide range of temperatures and pressures, thus demonstrating the accuracy and predictive power of the polarizable potentials used herein. The theoretical isosteric heat of adsorption (Qst) values are also in good agreement with the newly reported experimental Qst values for the respective sorbates in UTSA-20. Sorption onto the more positively charged Cu2+ ion of the [Cu2(O2CR)4] cluster was observed for both CO2 and H2. However, a binding site with energetics comparable to that for an open-metal site was also discovered for both sorbates. A radial distribution function (g(r)) analysis about the preferential Cu2+ ions for CO2 and H2 revealed that both sorbates display different trends for the relative occupancy about such sites upon increasing/decreasing the pressure in the MOF. Overall, this study provides insights into the CO2 and H2 sorption mechanisms in this MOF containing open-metal sites and small pore sizes for the first time through a classical polarizable force field.
 Please wait while we load your content...
Please wait while we load your content...

