
Linear-scaling density functional simulations of the effect of crystallographic structure on the electronic and optical properties of fullerene solvates†
 ab
ab
Abstract
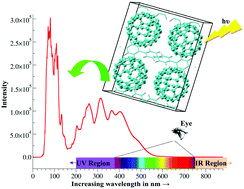
In this work, the crystal properties, HOMO and LUMO energies, band gaps, density of states, as well as the optical absorption spectra of fullerene C60 and its derivative phenyl-C61-butyric-acid-methyl-ester (PCBM) co-crystallised with various solvents such as benzene, biphenyl, cyclohexane, and chlorobenzene were investigated computationally using linear-scaling density functional theory with plane waves as implemented in the ONETEP program. Such solvates are useful materials as electron acceptors for organic photovoltaic (OPV) devices. We found that the fullerene parts contained in the solvates are unstable without solvents, and the interactions between fullerene and solvent molecules in C60 and PCBM solvates make a significant contribution to the cohesive energies of solvates, indicating that solvent molecules are essential to keep C60 and PCBM solvates stable. Both the band gap (Eg) and the HOMO and LUMO states of C60 and PCBM solvates are mainly determined by the fullerene parts contained in solvates. Chlorobenzene- and ortho-dichlorobenzene-solvated PCBM are the most promising electron-accepting materials among these solvates for increasing the driving force for charge separation in OPVs due to their relatively high LUMO energies. The UV-Vis absorption spectra of solvent-free C60 and PCBM crystals in the present work are similar to those of C60 and PCBM thin films shown in the literature. Changes in the absorption spectra of C60 solvates relative to the solvent-free C60 crystal are more significant than those of PCBM solvates due to the weaker effect of solvents on the π-stacking interactions between fullerene molecules in the latter solvates. The main absorptions for all C60 and PCBM crystals are located in the ultraviolet (UV) region.
 Please wait while we load your content...
Please wait while we load your content...

