
A molecular dynamics study of the interaction of water with the external surface of silicalite-1†

Abstract
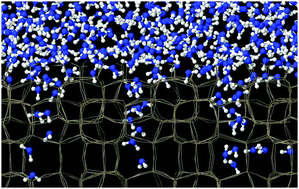
The interaction of water with the hydroxylated (010) surface of silicalite-1 was studied by classical molecular dynamics simulations. Interatomic interactions in the system were described using a set of effective potentials combining well-tested BKS and SPC models. The extended force field is shown to correctly reproduce the structural, energy, and dynamical characteristics of the silica surface OH groups. The interaction of water with the hydrophilic silanols leads to an ordering of H2O molecules in the vicinity of the surface. The ordering is found to be limited to two molecular layers extending to 7 Å above the surface. Despite the hydrophobic nature of the silicalite structure and the presence of hydrophilic surface sites, water molecules are capable of penetrating the porous silicalite system, where they form an H-bonded network blocking further access to the bulk. Water uptake by the zeolite was computed to be small in the time-scale of the simulations. The vibrational dynamics of the surface OH groups and adsorbed water molecules is discussed in detail. In agreement with the results of spectroscopic experiments, water molecules in the ordered surface layer have a spectral signature different from that of molecules more distant from the surface.
 Please wait while we load your content...
Please wait while we load your content...

