
Mapping the sequence–structure relationships of simple cyclic hexapeptides†
 *a
*a
Abstract
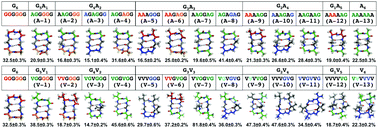
Cyclic peptides are promising protein–protein interaction modulators with high binding affinities and specificities, as well as enhanced stabilities and oral availabilities over linear analogs. Despite their relatively small size and cyclic architecture, it is currently difficult to predict the favored conformation(s) of most classes of cyclic peptides. An improved understanding of the sequence–structure relationships for cyclic peptides will offer an avenue for the rational design of cyclic peptides as possible therapeutics. In this work, we systematically explored the sequence–structure relationships for two cyclic hexapeptide systems using molecular dynamics simulation techniques. Starting with an all-glycine cyclic hexapeptide, cyclo-G6, we systematically replaced glycine residues with alanines and characterized the structural ensembles of different variants. The same process was repeated with valines to investigate the effects of larger side chains. An analysis of the origin of structure preferences was performed using thermodynamics decomposition and several general observations are reported.
 Please wait while we load your content...
Please wait while we load your content...

