
Evaluating the electronic structure of formal LnII ions in LnII(C5H4SiMe3)31− using XANES spectroscopy and DFT calculations†
 ‡a
Maryline G.
Ferrier,
‡b
Jing
Su,
‡b
Enrique
Batista,
*b
Samantha K.
Cary,
b
Jonathan W.
Engle,
bc
William J.
Evans,
*a
Juan S.
Lezama Pacheco,
d
Stosh A.
Kozimor,
*b
Angela C.
Olson,
b
Austin J.
Ryan,
a
Benjamin W.
Stein,
b
Gregory L.
Wagner,
b
David H.
Woen,
a
Ping
Yang
*b
‡a
Maryline G.
Ferrier,
‡b
Jing
Su,
‡b
Enrique
Batista,
*b
Samantha K.
Cary,
b
Jonathan W.
Engle,
bc
William J.
Evans,
*a
Juan S.
Lezama Pacheco,
d
Stosh A.
Kozimor,
*b
Angela C.
Olson,
b
Austin J.
Ryan,
a
Benjamin W.
Stein,
b
Gregory L.
Wagner,
b
David H.
Woen,
a
Ping
Yang
*b
Abstract
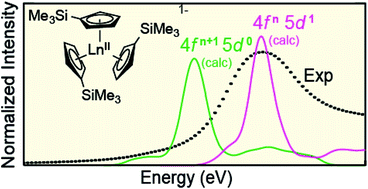
The isolation of [K(2.2.2-cryptand)][Ln(C5H4SiMe3)3], formally containing LnII, for all lanthanides (excluding Pm) was surprising given that +2 oxidation states are typically regarded as inaccessible for most 4f-elements. Herein, X-ray absorption near-edge spectroscopy (XANES), ground-state density functional theory (DFT), and transition dipole moment calculations are used to investigate the possibility that Ln(C5H4SiMe3)31− (Ln = Pr, Nd, Sm, Gd, Tb, Dy, Y, Ho, Er, Tm, Yb and Lu) compounds represented molecular LnII complexes. Results from the ground-state DFT calculations were supported by additional calculations that utilized complete-active-space multi-configuration approach with second-order perturbation theoretical correction (CASPT2). Through comparisons with standards, Ln(C5H4SiMe3)31− (Ln = Sm, Tm, Yb, Lu, Y) are determined to contain 4f6 5d0 (SmII), 4f13 5d0 (TmII), 4f14 5d0 (YbII), 4f14 5d1 (LuII), and 4d1 (YII) electronic configurations. Additionally, our results suggest that Ln(C5H4SiMe3)31− (Ln = Pr, Nd, Gd, Tb, Dy, Ho, and Er) also contain LnII ions, but with 4fn 5d1 configurations (not 4fn+1 5d0). In these 4fn 5d1 complexes, the C3h-symmetric ligand environment provides a highly shielded 5d-orbital of a′ symmetry that made the 4fn 5d1 electronic configurations lower in energy than the more typical 4fn+1 5d0 configuration.
 Please wait while we load your content...
Please wait while we load your content...

