
Mechanism and reactivity in the Morita–Baylis–Hillman reaction: the challenge of accurate computations†
 *a
and
Raghavan B.
Sunoj
*c
*a
and
Raghavan B.
Sunoj
*c
Abstract
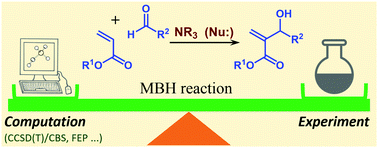
A systematic density functional theory exploration of various reactive steps together with benchmark coupled cluster results are used to propose an accurate model of the mechanism of the Morita–Baylis–Hillman (MBH) reaction in organic chemistry. This reaction has attracted considerable interest from the synthetic and mechanistic points of view in recent years, with both computational and experimental mechanistic studies. It has recently (R. E. Plata and D. A. Singleton, J. Am. Chem. Soc., 2015, 137, 3811–3826) been correctly pointed out that previous computational studies failed to reproduce known mechanistic features of the reaction. The same study argued that computation is simply not able at the present time to provide accurate models for such reactions. This second claim is shown by our present work to overstate the problem: by using current ‘state of the art’ methodology, our results are fully consistent with observed behavior within the expected error bars of 1–5 kcal mol−1, far smaller than the errors reported in Plata and Singleton's study. On the basis of exhaustive calculations reported here, we suggest that our proposed approaches for modeling electronic structure, solvation, and entropy should be able to provide accurate predictions for many more reactions. We also suggest that reactions like the MBH reaction, where solvation and entropy effects are particularly large, are among the hardest for computational mechanistic studies.
 Please wait while we load your content...
Please wait while we load your content...

