
Computational investigations of electronic structure modifications of ferrocene-terminated self-assembled monolayers: effects of electron donating/withdrawing functional groups attached on the ferrocene moiety†
 *a
Yoshitada
Morikawa
*cd
and
*a
Yoshitada
Morikawa
*cd
and
Abstract
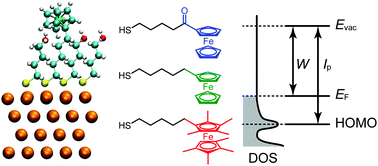
The electrochemical properties of chemically modified electrodes have long been a significant focus of research. Although the electronic states are directly related to the electrochemical properties, there have been only limited systematic efforts to reveal the electronic structures of adsorbed redox molecules with respect to the local environment of the redox center. In this study, density functional theory (DFT) calculations were performed for ferrocene-terminated self-assembled monolayers with different electron-donating abilities, which can be regarded as the simplest class of chemically modified electrodes. We revealed that the local electrostatic potentials, which are changed by the electron donating/withdrawing functional groups at the ferrocene moiety and the dipole field of coadsorbed inert molecules, practically determine the density of states derived from the highest occupied molecular orbital (HOMO) and its vicinities (HOMO−1 and HOMO−2) with respect to the electrode Fermi level. Therefore, to design new, sophisticated electrodes with chemical modification, one should consider not only the electronic properties of the constituent molecules, but also the local electrostatic potentials formed by these molecules and coadsorbed inert molecules.
 Please wait while we load your content...
Please wait while we load your content...

