
Unraveling innate substrate control in site-selective palladium-catalyzed C–H heterocycle functionalization†
Abstract
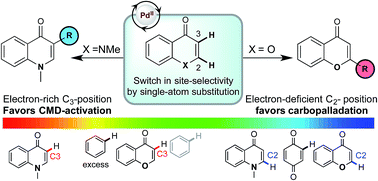
Understanding the regioselectivity of C–H activation in the absence of directing groups is an important step towards the design of site-selective C–H functionalizations. The Pd(II)-catalyzed direct arylation of chromones and enaminones provides an intriguing example where a simple substitution leads to a divergence in substrate-controlled site-selectivity. We describe computational and experimental studies which reveal this results from a switch in mechanism and therefore the selectivity-determining step. We present computational results and experimentally measured kinetic isotope effects and labelling studies consistent with this proposal. The C–H activation of these substrates proceeds via a CMD mechanism, which favors more electron rich positions and therefore displays a pronounced kinetic selectivity for the C3-position. However, C2-selective carbopalladation is also a competitive pathway for chromones so that the overall regiochemical outcome depends on which substrate undergoes activation first. Our studies provide insight into the site-selectivity based on the favorability of two competing CMD and carbopalladation processes of the substrates undergoing coupling. This model can be utilized to predict the regioselectivity of coumarins which are proficient substrates for carbopalladation. Furthermore, our model is able to account for the opposite selectivities observed for enaminone and chromone, and explains how a less reactive coupling partner leads to a switch in selectivity.
 Please wait while we load your content...
Please wait while we load your content...

