
Graphyne and graphdiyne: theoretical insight into ground and excited state properties

Abstract
The two carbon allotropes, graphyne (C66H18) (gr1) and graphdiyne (C90H18) (gr2), in the form of nanoflakes were studied with the help of density functional theory (DFT) and time dependent density functional theory (TDDFT). The basis sets used for DFT and TDDFT calculations were 6-31+G(d) and 6-31+G(d, p). The LSDA functional was found to be more appropriate for describing the electronic and optical properties, as compared to the B3LYP functional. Electronic gaps, binding energy, dipole moments and bond lengths were determined. Both structures were found to be semiconducting in nature. Furthermore, the absorption process was thoroughly studied along with excited state analysis, which includes the oscillator strengths, natural transition orbital (NTO) analysis, exciton size and electron–hole (e–h) correlation plots. The “gr1” absorption range (1.59–3.59 eV) was found to be wider than that of “gr2” (1.91–2.49 eV). From NTO analysis, electron delocalization in “gr2” is found to be more than that in “gr1” during absorption. The nature of the excitons in both structures is Frenkel, indicating a higher exciton binding energy. The charge transfer mechanism was studied using e–h correlation plots and was found to more diverse in “gr2” than “gr1”.
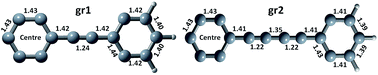
 Please wait while we load your content...
Please wait while we load your content...

