
DFT study on the iridium-catalyzed multi-alkylation of alcohol with ammonia†
Abstract
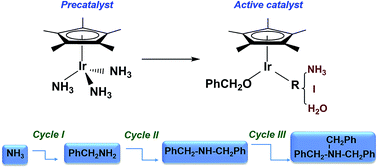
The catalytic mechanism for the multi-alkylation of benzyl alcohols with ammonia catalyzed by the water-soluble catalyst, [Cp*IrIII(NH3)3][I]2, is computationally investigated by density functional theory (DFT). Three possible active catalysts based on various ligands, NH3, I, and H2O were investigated for the first catalytic cycle and the active catalyst with an ammine ligand is the best one. For the three interrelated and succession catalytic cycles (I, II, and III), each catalytic cycle consists of three stages: (stage I) the oxidation of the alkoxide ligand to free benzaldehyde; (stage II) the dehydration coupling between benzaldehyde and amine-reactant without the participation of the metal catalyst; (stage III) the reduction of imine by a hydride intermediate to give an amine-product. The three catalytic cycles have the same stage I, where the β-H elimination step is the highest point of the profile. The transfer hydrogenation step of stage III is believed to be the rate-determining step of every catalytic cycle. Remarkably, for the three catalytic cycles, the active barrier of the key reductive hydride transfer gradually increases due to both electric and steric effects induced by the adding of the benzyl ring of the amine-reactant. Our computational results are in good agreement with the experimental outcomes of the reaction.
 Please wait while we load your content...
Please wait while we load your content...

