
Water oxidation catalysis – role of redox and structural dynamics in biological photosynthesis and inorganic manganese oxides†
Abstract
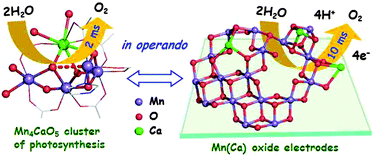
Water oxidation is pivotal in biological photosynthesis, where it is catalyzed by a protein-bound metal complex with a Mn4Ca-oxide core; related synthetic catalysts may become key components in non-fossil fuel technologies. Going beyond characterization of the catalyst resting state, we compare redox and structural dynamics of three representative birnessite-type Mn(Ca) oxides (catalytically active versus inactive; with/without calcium) and the biological catalyst. In the synthetic oxides, Mn oxidation was induced by increasingly positive electrode potentials and monitored by electrochemical freeze-quench and novel time-resolved in situ experiments involving detection of X-ray absorption and UV-vis transients, complemented by electrochemical impedance spectroscopy. A minority fraction of Mn(III) ions present at catalytic potentials is found to be functionally crucial; calcium ions are inessential but tune redox properties. Redox-state changes of the water-oxidizing Mn oxide are similarly fast as observed in the biological catalyst (<10 ms), but 10–100 times slower in the catalytically inactive oxide. Surprisingly similar redox dynamics of biological catalyst and water-oxidizing Mn(Ca) oxides suggest that in both catalysts, rather than direct oxidation of bound water species, oxidation equivalents are accumulated before onset of the multi-electron O–O bond formation chemistry in Mn(III)–Mn(IV) oxidation steps coupled to changes in the oxo-bridging between metal ions. Aside from the ability of the bulk oxide to undergo Mn oxidation-state changes, we identify two further, likely interrelated prerequisites for catalytic activity of the synthetic oxides: (i) the presence of Mn(III) ions at catalytic potentials preventing formation of an inert all-Mn(IV) oxide and (ii) fast rates of redox-state changes approaching the millisecond time domain.
 Please wait while we load your content...
Please wait while we load your content...

