
Structural and mechanistic basis for the high activity of Fe–N–C catalysts toward oxygen reduction†
Abstract
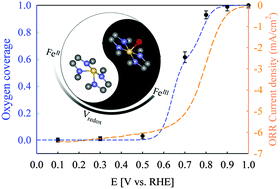
The development of efficient non-platinum group metal (non-PGM) catalysts for oxygen reduction reaction (ORR) is of paramount importance for clean and sustainable energy storage and conversion devices. The major bottleneck in developing Fe–N–C materials as the leading non-PGM catalysts lies in the poor understanding of the nature of active sites and reaction mechanisms. Herein, we report a scalable metal organic framework-derived Fe–N–C catalyst with high ORR activity demonstrated in practical H2/air fuel cells, and an unprecedented turnover frequency (TOF) in acid in rotating disk electrode. By characterizing the catalyst under both ex situ and operando conditions using combined microscopic and spectroscopic techniques, we show that the structures of active sites under ex situ and working conditions are drastically different. Resultantly, the active site proposed here, a non-planar ferrous Fe–N4 moiety embedded in distorted carbon matrix characterized by a high Fe2+/3+ redox potential, is in contrast with those proposed hitherto derived from ex situ characterizations. This site reversibly switches to an in-plane ferric Fe–N4 moiety poisoned by oxygen adsorbates during the redox transition, with the population of active sites controlled by the Fe2+/3+ redox potential. The unprecedented TOF of the active site is correlated to its near-optimal Fe2+/3+ redox potential, and essentially originated from its favorable biomimetic dynamic nature that balances the site-blocking effect and O2 dissociation. The porous and disordered carbon matrix of the catalyst plays pivotal roles for its measured high ORR activity by hosting high population of reactant-accessible active sites.
 Please wait while we load your content...
Please wait while we load your content...

