
The significance of secondary interactions during alkaline earth-promoted dehydrogenation of dialkylamine-boranes†
Abstract
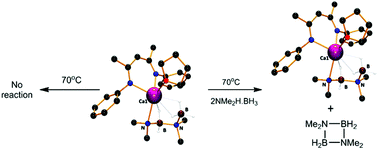
Reactions of anilidoimine magnesium n-butyl and calcium bis(trimethylsilyl)amide derivatives with Me2NH·BH3 at 25 °C resulted in the isolation of complexes containing [NMe2BH2NMe2BH3]− and [NMe2BH3]− anions respectively. Although onward reaction of the calcium species at 30 °C with a further equivalent of Me2NH·BH3 provided ca. 90% conversion of the coordinated dimethylamidoborane anion to [NMe2BH2NMe2BH3]−, this process also resulted in significant (ca. 25%) levels of competitive protonation of the anilidoimine spectator ligand. A similar reaction performed between a previously reported β-diketiminato calcium dimethylamidoborane and Me2NH·BH3, however, provided clean conversion to a structurally characterised calcium [NMe2BH2NMe2BH3]− complex. Reaction of a more sterically congested β-diketiminato magnesium n-butyl reagent with Me2NH·BH3 has allowed the isolation of a magnesium derivative of the [NMe2BH3]− anion. The thermal stability of these compounds as well as previously reported magnesium and calcium amidoborane species indicate, in partial agreement with a recent DFT study, that all of these compounds are resistant to the β- and δ-hydride elimination reactions that have previously been implicated as the key B–N bond-forming and dehydrogenative steps in the group 2-catalysed dehydrocoupling of Me2NH·BH3. In contrast to these observations, addition of stoichiometric quantities of Me2NH·BH3 to the various isolated group 2 amidoborane species was found to result in facile elimination of the cyclic borazane [Me2N–BH2]2 which occurs with regeneration of the metallated amidoborane. On this basis, we suggest that the dehydrocoupling of Me2NH·BH3 at group 2 centres takes place as a sequence of concerted proton-assisted steps during which B–H and N–H bond breaking plays an equally prominent role, with the efficacy of boron hydride protonolysis dictated by the relative polarising influence of the B–H to Mg/Ca interactions. Furthermore, we propose a modified mechanism for group 2-mediated dimethylamine borane dehydrocoupling that is dependent on the intermediacy of key derivatives of the [NMe2·BH3]− and [NMe2BH2NMe2BH3]− anions but does not require the formation of high energy alkaline earth hydride intermediates. Although these results are specifically focussed on the applications of alkaline earth species, this mechanistic insight may also be relevant to other redox-inactive main group element-based systems and to our understanding of hydrogen evolution from saline derivatives of ammonia borane.
 Please wait while we load your content...
Please wait while we load your content...

