
Theoretical prediction of coordination environments and stability constants of lanthanum lactate complexes in solution†
Abstract
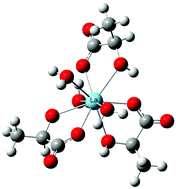
Using Density Functional Theory calculations in combination with explicit solvent and a continuum solvent model, this work sets out to understand the coordination environment and relevant thermodynamics of La(III)-lactate complexes. Calculations focus on the coordination modes for the complexes and changes in Gibbs free energy for complexation in solution. These results confirm that the α-hydroxyl group should be protonated, or at least hydrogen bonded to a water molecule, upon successive addition of the lactate ligand to the La(III) center using Bader's Atoms-in Molecules (AIM) approach. In addition, we present a straightforward method for predicting stability constants at the semi-quantitative level for La(III)-lactate complexes in solution. The proposed method could be particularly useful for prediction of lanthanide complex formation in various biochemical, environmental, and nuclear separations processes.
 Please wait while we load your content...
Please wait while we load your content...

