
Time-resolved signatures across the intramolecular response in substituted cyanine dyes†
 a
a
Abstract
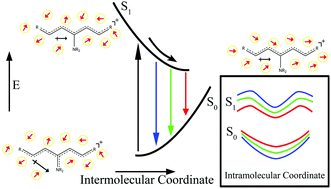
The optically populated excited state wave packet propagates along multidimensional intramolecular coordinates soon after photoexcitation. This action occurs alongside an intermolecular response from the surrounding solvent. Disentangling the multidimensional convoluted signal enables the possibility to separate and understand the initial intramolecular relaxation pathways over the excited state potential energy surface. Here we track the initial excited state dynamics by measuring the fluorescence yield from the first excited state as a function of time delay between two color femtosecond pulses for several cyanine dyes having different substituents. We find that when the high frequency pulse precedes the low frequency one and for timescales up to 200 fs, the excited state population can be depleted through stimulated emission with efficiency that is dependent on the molecular electronic structure. A similar observation at even shorter times was made by scanning the chirp (frequencies ordering) of a femtosecond pulse. The changes in depletion reflect the rate at which the nuclear coordinates of the excited state leave the Franck–Condon (FC) region and progress towards achieving equilibrium. Through functional group substitution, we explore these dynamic changes as a function of dipolar change following photoexcitation. Density functional theory calculations were performed to provide greater insight into the experimental spectroscopic observations. Complete active space (CAS) self-consistent field and CAS second order perturbation theory calculated potential energy surfaces tracking twisting and pyramidalization confirm that the steeper potential at the FC region leads to the observation of faster wave packet dynamics.
 Please wait while we load your content...
Please wait while we load your content...

