
Requirements of first-principles calculations of X-ray absorption spectra of liquid water†

Abstract
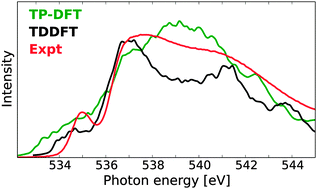
A computational benchmark study on X-ray absorption spectra of water has been performed by means of transition-potential density functional theory (TP-DFT), damped time-dependent density functional theory (TDDFT), and damped coupled cluster (CC) linear response theory. For liquid water, using TDDFT with a tailored CAM-B3LYP functional and a polarizable embedding, we find that an embedding with over 2000 water molecules is required to fully converge spectral features for individual molecules, but a substantially smaller embedding can be used within averaging schemes. TP-DFT and TDDFT calculations on 100 MD structures demonstrate that TDDFT produces a spectrum with spectral features in good agreement with experiment, while it is more difficult to fully resolve the spectral features in the TP-DFT spectrum. Similar trends were also observed for calculations of bulk ice. In order to further establish the performance of these methods, small water clusters have been considered also at the CC2 and CCSD levels of theory. Issues regarding the basis set requirements for spectrum simulations of liquid water and the determination of gas-phase ionization potentials are also discussed.
 Please wait while we load your content...
Please wait while we load your content...

