
The role of solvent in the self-assembly of m-aminobenzoic acid: a density functional theory and molecular dynamics study†
Abstract
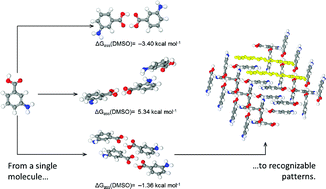
Solvent can have significant effects on the solution thermodynamics and crystallisation kinetics of organic compounds from solution. In the present work, the early stages of aggregation of the organic molecule m-aminobenzoic (mABA) in two different solvents, dimethyl sulfoxide (DMSO) and water were studied using a combination of quantum chemistry, molecular dynamics and metadynamics simulations. Density functional theory (B97-D and M06-2X) calculations with the continuum solvation SMD model were used to probe the potential energy surface of molecular clusters of m-aminobenzoic acid, (mABA)n (n = 2–4), locate their low-lying energy structures, and compute the Gibbs free energies of (mABA)n in solution. Starting from a large number of randomly generated candidate structures and by imposing the condition of minimum free energy in solution for the isomers of (mABA)n, we proved that the most stable dimer and tetramer in solution correspond to the classic carboxylic dimer π–π stacking synthon found in the crystalline form-II of mABA. Molecular dynamics simulations of mABA solutions at different concentrations reveal a significant solvent-dependent aggregation behaviour for mABA: in water, even at low concentrations, mABA molecules spontaneously form H-bonded π–π stacking molecular clusters, whereas in organosulfur solutions the molecules of mABA are in a more solvated state. Metadynamics simulations and microsolvation density functional theory calculations rationalize the low level of mABA aggregation in DMSO in terms of the energetic barrier for the desolvation of mABA molecules and formation of dimers, and the strength of mABA-solvent interactions, which are both larger in DMSO compared with water. This work shows that the solvent and its specific interaction with the organic solute molecules influences both the thermodynamics and kinetics of the molecular self-assembly process.
 Please wait while we load your content...
Please wait while we load your content...

