
Ab initio static and dynamic study of CH3NH3PbI3 degradation in the presence of water, hydroxyl radicals, and hydroxide ions†
Abstract
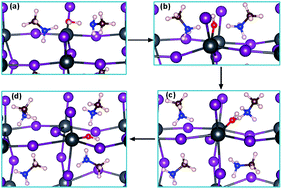
Perovskite solar cells have attracted great attention in the past few years due to their many advantages like superb photovoltaic and electronic properties, simple fabrication procedures and low price of raw materials. However, numerous studies have demonstrated that the perovskite materials degrade rapidly under humid air and sunlight, which greatly hinders perovskite solar cells from practical application. In this paper, density functional theory (DFT) calculations are combined with the ab initio molecular dynamics (AIMD) and climbing image nudged elastic band (CI-NEB) techniques to unveil the microscopic details of the degradation of CH3NH3PbI3 (MAPbI3). DFT calculations show that bulk MAPbI3 reacts readily with water molecules to form a monohydrate MAPbI3·H2O phase. However, subsequent degradation of this monohydrate is energetically unfavorable. On the other hand, in the presence of hydroxyl or hydroxide species, we identify possible pathways that lead to the full decomposition of MAPbI3 to the experimentally-identified degradation products. Furthermore, AIMD simulations were carried out to study the dynamics of the interaction of water molecules, hydroxyl radicals and hydroxide ions with the (110) surface of MAPbI3. Hydrogen transfer (or proton transfer) between the OH (or OH−) species and the CH3NH3 cation of the MAPbI3 surface was observed within the first 1 to 2 ps. Lastly, we studied the possible infiltration of H2O molecules, OH radicals and OH− ions in the (110) surface of MAPbI3 using the CI-NEB technique. The energy barriers for the infiltration processes in the three cases were found to be comparable.
 Please wait while we load your content...
Please wait while we load your content...

