
An ab initio and DFT study of the autoxidation of THF and THP†
Abstract
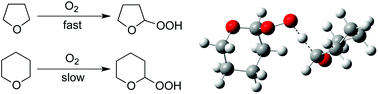
Tetrahydropyran (THP) is known to undergo autoxidation much more slowly than tetrahydrofuran (THF). To investigate the difference in reactivity in the autoxidation of these two ethers, ab initio and DFT calculations were carried out. At the BHandHLYP/aug-cc-pVDZ//BHandHLYP/cc-pVDZ level of theory, the energy barrier for hydrogen abstraction from THP is predicted to be 104.1 kJ mol−1, whereas that for THF is calculated as 94.1 kJ mol−1. Including solvation effects in the calculations lowers these barriers to 98.0 (THP) and 84.4 kJ mol−1 (THF); the energy barrier for the process involving THP is smaller by 14 kJ mol−1 than that for THF. While scanning the potential energy surface for the radical coupling process between the THP (or THF) radical with molecular oxygen, an energy barrier of 11.2 kJ mol−1 (BHandHLYP/6-311G**) was found for the process involving the THP radical, although no barrier was found for the reaction involving THF. Analysis of the Kohn–Sham singly occupied molecular orbitals (SOMOs) in the hydroperoxy complexes reveals that the SOMO of the THP complex would be blocked by the neighbouring hydrogen atoms in the THP ring. These factors would work together to delay the autoxidation of THP.
 Please wait while we load your content...
Please wait while we load your content...

