
Rotational spectroscopy of methyl benzoylformate and methyl mandelate: structure and internal dynamics of a model reactant and product of enantioselective reduction†
Abstract
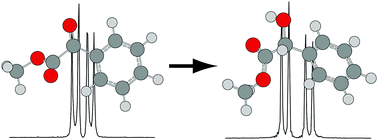
Pure rotational spectra of a prototypical prochiral ester, methyl benzoylformate (MBF), and the product of its enantioselective reduction, (R)-(−)-methyl mandelate (MM), were measured in the range of 5–16 GHz, using a cavity-based molecular beam Fourier-transform microwave spectrometer. Potential conformers were located using density functional theory calculations, and one conformer of each species was identified experimentally. The minimum energy conformer of MBF, in which the ester group is in a Z orientation, was observed for the first time. Based on an atoms-in-molecules analysis, MBF contains a weak CH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bond between the carbonyl oxygen atom of the ester group and the nearest hydrogen atom of the aromatic ring. In the minimum energy conformer of MM, the ester group is oriented to accommodate a hydrogen bond between the hydrogen atom of the hydroxyl group and the carbonyl oxygen atom (OH⋯OC), rather than the sp3 oxygen atom (OH⋯O–C). For both species, splittings of the rotational transitions were observed, which are attributed to methyl internal rotation, and the orientations and barrier heights of the methyl tops were determined precisely. The barrier heights for MBF and MM are 4.60(2) and 4.54(3) kJ mol−1, respectively, which are consistent with values predicted by high-level wavefunction-based calculations. On the basis of an atoms-in-molecules analysis, we propose that destabilization of the sp3 oxygen atom of the ester group most directly dictates the barrier height.
C hydrogen bond between the carbonyl oxygen atom of the ester group and the nearest hydrogen atom of the aromatic ring. In the minimum energy conformer of MM, the ester group is oriented to accommodate a hydrogen bond between the hydrogen atom of the hydroxyl group and the carbonyl oxygen atom (OH⋯OC), rather than the sp3 oxygen atom (OH⋯O–C). For both species, splittings of the rotational transitions were observed, which are attributed to methyl internal rotation, and the orientations and barrier heights of the methyl tops were determined precisely. The barrier heights for MBF and MM are 4.60(2) and 4.54(3) kJ mol−1, respectively, which are consistent with values predicted by high-level wavefunction-based calculations. On the basis of an atoms-in-molecules analysis, we propose that destabilization of the sp3 oxygen atom of the ester group most directly dictates the barrier height.
 Please wait while we load your content...
Please wait while we load your content...

