
Vibrational energy transfer dynamics in ruthenium polypyridine transition metal complexes
Abstract
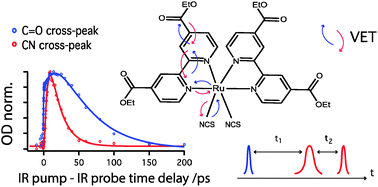
Understanding the dynamics of the initial stages of vibrational energy transfer in transition metal complexes is a challenging fundamental question which is also of crucial importance for many applications, such as improving the performance of solar devices or photocatalysis. The present study investigates vibrational energy transport in the ground and the electronic excited state of Ru(4,4′-(COOEt)2-2,2-bpy)2(NCS)2, a close relative of the efficient “N3” dye used in dye-sensitized solar cells. Using the emerging technique of ultrafast two-dimensional infrared spectroscopy, we show that, similarly to other transition-metal complexes, the central Ru heavy atom acts as a “bottleneck” making the energy transfer from small ligands with high energy vibrational stretching frequencies less favorable and thereby affecting the efficiency of vibrational energy flow in the complex. Comparison of the vibrational relaxation times in the electronic ground and excited state of Ru(4,4′-(COOEt)2-2,2-bpy)2(NCS)2 shows that it is dramatically faster in the latter. We propose to explain this observation by the intramolecular electrostatic interactions between the thiocyanate group and partially oxidised Ru metal center, which increase the degree of vibrational coupling between CN and Ru–N modes in the excited state thus reducing structural and thermodynamic barriers that slow down vibrational relaxation and energy transport in the electronic ground state. As a very similar behavior was earlier observed in another transition-metal complex, Re(4,4′-(COOEt)2-2,2′-bpy)(CO)3Cl, we suggest that this effect in vibrational energy dynamics might be common for transition-metal complexes with heavy central atoms.
 Please wait while we load your content...
Please wait while we load your content...

