
Static and lattice vibrational energy differences between polymorphs†

Abstract
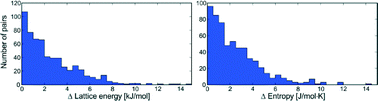
A computational study of 1061 experimentally determined crystal structures of 508 polymorphic organic molecules has been performed with state-of-the-art lattice energy minimisation methods, using a hybrid method that combines density functional theory intramolecular energies with an anisotropic atom–atom intermolecular model. Rigid molecule lattice dynamical calculations have also been performed to estimate the vibrational contributions to lattice free energies. Distributions of the differences in lattice energy, free energy, zero point energy, entropy and heat capacity between polymorphs are presented. Polymorphic lattice energy differences are typically very small: over half of polymorph pairs are separated by less than 2 kJ mol−1 and lattice energy differences exceed 7.2 kJ mol−1 in only 5% of cases. Unsurprisingly, vibrational contributions to polymorph free energy differences at ambient conditions are dominated by entropy differences. The distribution of vibrational energy differences is narrower than lattice energy differences, rarely exceeding 2 kJ mol−1. However, these relatively small vibrational free energy contributions are large enough to cause a re-ranking of polymorph stability below, or at, room temperature in 9% of the polymorph pairs.
- This article is part of the themed collections: A celebration of 25 volumes of CrystEngComm, Editor’s Collection: Polymorphism in Molecular Crystals and Polymorphism
 Please wait while we load your content...
Please wait while we load your content...

