
Combined experimental–theoretical study of the optoelectronic properties of non-stoichiometric pyrochlore bismuth titanate†
Abstract
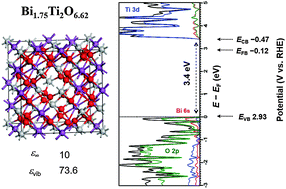
A combination of experimental and computational methods was applied to investigate the crystal structure and optoelectronic properties of the non-stoichiometric pyrochlore Bi2−xTi2O7−1.5x. The detailed experimental protocol for both powder and thin-film material synthesis revealed that a non-stoichiometric Bi2−xTi2O7−1.5x structure with an x value of ∼0.25 is the primary product, consistent with the thermodynamic stability of the defect-containing structure computed using density functional theory (DFT). The approach of density functional perturbation theory (DFPT) was used along with the standard GGA PBE functional and the screened Coulomb hybrid HSE06 functional, including spin–orbit coupling, to investigate the electronic structure, the effective electron and hole masses, the dielectric constant, and the absorption coefficient. The calculated values for these properties are in excellent agreement with the measured values, corroborating the overall analysis. This study indicates potential applications of bismuth titanate as a wide-bandgap material, e.g., as a substitute for TiO2 in dye-sensitized solar cells and UV-light-driven photocatalysis.
 Please wait while we load your content...
Please wait while we load your content...

