
Structural investigations by in silico modeling for designing NR2B subunit selective NMDA receptor antagonists
Abstract
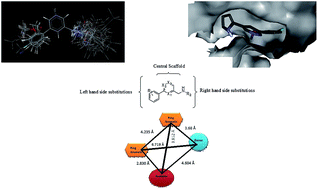
Glutamate N-methyl-D-aspartate (NMDA) receptors are widely distributed throughout the mammalian central nervous system (CNS). They play key roles in many brain disorders, such as brain trauma, seizures, pain, Parkinson's and Huntington's disease. NMDA receptors are co-assemblies of four subunits. The selectivity of ligands for the NR2B subunit, out of the others, has shown the best side effect profile. The objective of the current studies is to establish the structural requirements for pyrazine and related derivatives for being NR2B selective NMDA receptor antagonists. 2D- and 3D-QSAR models have been developed. 2D-QSAR analyses revealed the key role of Baumann's alignment independent topological descriptors, such as T_2_F_7, T_C_O_7 and T_T_T_6, in biological activity prediction. Furthermore, 3D-QSAR analyses showed that steric and electrostatic molecular descriptors have significant influence in the optimization of pyrazine and related lead compounds as NR2B selective NMDA receptor antagonists. 3D points generated using a 3D-QSAR study have been studied to uncover minimum structural requirements of pyrazine derivatives. The significantly high prediction power of the best QSAR model supports the mechanism of NMDA receptor antagonism through NR2B subunit selectivity. A molecular docking study of the most active compound 18 revealed important structural insights needed to optimize pyrazine derivatives for use as NR2B selective NMDA receptor antagonists. Moreover, a putative structure-based pharmacophore model was established, having features of two aromatic rings, one hydrogen bond donor and one hydrogen bond acceptor. This pharmacophore model could be used as a query for virtual screening of commercially available chemical databases to identify new hits, and 2D- and 3D-QSAR models could be used to predict the activities of identified hits.
 Please wait while we load your content...
Please wait while we load your content...

