
Tug-of-war between corrugation and binding energy: revealing the formation of multiple moiré patterns on a strongly interacting graphene–metal system†

Abstract
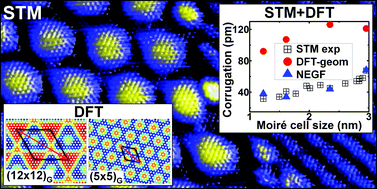
The formation of multidomain epitaxial graphene on Rh(111) under ultra-high vacuum (UHV) conditions has been characterized by scanning tunnelling microscopy (STM) measurements and density functional theory (DFT) calculations. At variance with the accepted view for strongly interacting graphene–metal systems, we clearly demonstrate the formation of different rotational domains leading to multiple moiré structures with a wide distribution of surface periodicities. Experiments reveal a correlation between the STM apparent corrugation and the lattice parameter of the moiré unit cell, with corrugations of just 30–40 pm for the smallest moirés. DFT calculations for a relevant selection of these moiré patterns show much larger height differences and a non-monotonic behaviour with the moiré size. Simulations based on non-equilibrium Green's function (NEGF) methods reproduce quantitatively the experimental trend and provide a detailed understanding of the interplay between electronic and geometric contributions in the STM contrast of graphene systems. Our study sheds light on the subtle energy balance among strain, corrugation and binding that drives the formation of the moiré patterns in all graphene/metal systems and suggests an explanation for the success of an effective model only based on the lattice mismatch. Although low values of the strain energy are a necessary condition, it is the ability of graphene to corrugate in order to maximize the areas of favourable graphene–metal interactions that finally selects the stable configurations.
 Please wait while we load your content...
Please wait while we load your content...

