
Solvation effects on the band edge positions of photocatalysts from first principles†

Abstract
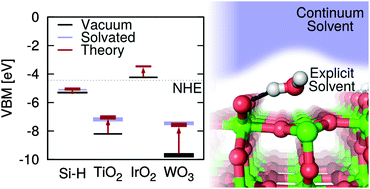
The band edge positions of photocatalysts relative to the redox potentials of water play an important role in determining the efficiency of photoelectrochemical cells. These band positions depend on the structure of the solid–liquid interface, but direct ab initio molecular dynamics calculations of these interfaces, while expected to be accurate, are too computationally demanding for high-throughput materials screening. Thus rapid theoretical screening of new photocatalyst materials requires simplified continuum solvation models that are suitable for treating solid–liquid interfaces. In this paper, we evaluate the accuracy of the recently developed CANDLE and SaLSA continuum solvation models for predicting solvation effects on the band positions of several well-studied surfaces [Si(111), TiO2(110), IrO2(110) and WO3(001)] in water. We find that the solvation effects vary considerably, ranging from <0.5 eV for hydrophobic surfaces, 0.5–1 eV for many hydrophilic oxide surfaces, to ∼2 eV for oxygen-deficient surfaces. The solvation model predictions are in excellent agreement (within ∼0.1 eV) with ab initio molecular dynamics results where available, and in good agreement (within ∼0.2–0.3 eV) with experimental measurements. We also predict the energetics for surface oxygen vacancies and their effect on the band positions of the hydrated WO3(001) surface, leading to an explanation for why the solvation shift observed experimentally is substantially larger than predicted for the ideal surface. Based on the correlation between solvation shift and the type of surface and solvent, we suggest approaches to engineer the band positions of surfaces in aqueous and non-aqueous solutions.
 Please wait while we load your content...
Please wait while we load your content...

