
Predicting stabilizing mutations in proteins using Poisson–Boltzmann based models: study of unfolded state ensemble models and development of a successful binary classifier based on residue interaction energies†
Abstract
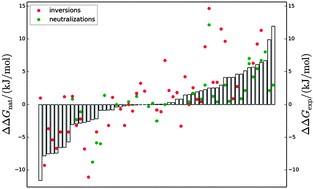
In many cases the stability of a protein has to be increased to permit its biotechnological use. Rational methods of protein stabilization based on optimizing electrostatic interactions have provided some fine successful predictions. However, the precise calculation of stabilization energies remains challenging, one reason being that the electrostatic effects on the unfolded state are often neglected. We have explored here the feasibility of incorporating Poisson–Boltzmann model electrostatic calculations performed on representations of the unfolded state as large ensembles of geometrically optimized conformations calculated using the ProtSA server. Using a data set of 80 electrostatic mutations experimentally tested in two-state proteins, the predictive performance of several such models has been compared to that of a simple one that considers an unfolded structure of non-interacting residues. The unfolded ensemble models, while showing correlation between the predicted stabilization values and the experimental ones, are worse than the simple model, suggesting that the ensembles do not capture well the energetics of the unfolded state. A more attainable goal is classifying potential mutations as either stabilizing or non-stabilizing, rather than accurately calculating their stabilization energies. To implement a fast classification method that can assist in selecting stabilizing mutations, we have used a much simpler electrostatic model based only on the native structure and have determined its precision using different stabilizing energy thresholds. The binary classifier developed finds 7 true stabilizing mutants out of every 10 proposed candidates and can be used as a robust tool to propose stabilizing mutations.
 Please wait while we load your content...
Please wait while we load your content...

