
Quantum chemical insights into the dependence of porphyrin basicity on the meso-aryl substituents: thermodynamics, buckling, reaction sites and molecular flexibility
Abstract
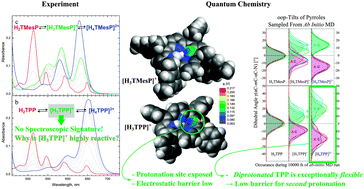
The chemical and sensing properties of porphyrins are frequently tuned via the introduction of peripheral substituents. In the context of the exceptionally fast second protonation step in the case of 5,10,15,20-tetraphenylporphyrin (TPP), as compared to porphin and 5,10,15,20-tetramesitylporphyrin (TMesP), we investigated the macrocycle-substituent interactions of these three porphyrin derivatives in detail. Using quantum chemical thermodynamics calculations, the analysis of geometric structures, torsional profiles, electrostatic potential distributions, and particularly the analysis of molecular flexibilities via ab initio molecular dynamics simulations, we obtained a comprehensive picture of the reactivities of the studied porphyrins and how these are influenced by the meso-substituents. As compared to porphin and TMesP the second protonation of TPP is energetically more favorable and is particularly energetically comparable to its first protonation, instead of being significantly less favorable like in the case of porphyrin and TMesP. Additionally, the second TPP protonation is facilitated by an interplay between out-of-plane (oop) distortion of the protonation site and a pronounced electrostatic binding spot at the protonation site. Furthermore, the second protonation is particularly facilitated in the case of TPP by the large oop-flexibility of the diprotonated species as unraveled by ab initio molecular dynamics simulations.
 Please wait while we load your content...
Please wait while we load your content...

