
Choosing a density functional for modeling adsorptive hydrogen storage: reference quantum mechanical calculations and a comparison of dispersion-corrected density functionals†
Abstract
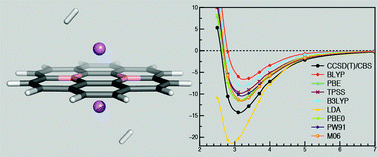
Hydrogen storage in carbonaceous materials and their derivatives is currently a widely investigated topic. The rational design of novel adsorptive materials is often attempted with the help of computational chemistry tools, in particular density functional theory (DFT). However, different exchange–correlation functionals provide a very wide range of hydrogen binding energies. The aim of this article is to offer high level QM reference data based on coupled-cluster singles and doubles calculations with perturbative triple excitations, CCSD(T), and a complete basis set limit estimate that can be used to assess the accuracy of various DFT-based predictions. For one complex, the CCSD(T) result is verified against diffusion quantum Monte Carlo calculations. Reference binding curves are calculated for two model compounds representing weak and strong hydrogen adsorption: coronene (−4.7 kJ mol−1 per H2), and coronene modified with boron and lithium (−14.3 kJ mol−1). The reference data are compared to results obtained with widely used density functionals including pure DFT, M06, DFT-D3, PBE-TS, PBE + MBD, optB88-vdW, vdW-DF, vdW-DF2 and VV10. We find that whereas DFT-D3 shows excellent results for weak hydrogen adsorption on coronene, most of the less empirical density based dispersion functionals except VV10 overestimate this interaction. On the other hand, some of the less empirical density based dispersion functionals better describe stronger binding in the more polar coroB2Li2⋯2H2 complex which is one of realistic models for high-capacity hydrogen storage materials. Our results may serve as a guide for choosing suitable DFT methods for quickly evaluating hydrogen binding potential and as a reference for assessing the accuracy of the previously published DFT results.
 Please wait while we load your content...
Please wait while we load your content...

